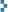
Plutonium is a practical and versatile substance, having applications that range from planetary extinction to routine fire protection, depending on the user’s fancy. The element has been mass-produced in nuclear reactors since World War Two, and occurs in various isotopic compositions in the discharged reactor fuel, reflecting variables such as fuel burnup, initial uranium enrichment, and neutron spectral characteristics of the reactor design. The Soviet Union cooked up more plutonium than any other nation. Most of this was slated for the noble purpose of containing capitalist imperialism, but some found its way into commercial ionization smoke detectors like the KI-1, RID-1, and RID-6M. (The bourgeois warmongers themselves preferred, and still prefer, americium-241 for this application.) Occasionally, people in the former USSR try to peddle their old smoke detector plutonium on the nuclear black market, thinking that it may attract top dollar from terrorists with an appetite for nuclear warfare. We’ll examine that possibility in more detail shortly.
Since I was curious about the technical characteristics of Soviet smoke detector plutonium, I picked up an old KI-1 smoke detector and sacked it for the source. The source design bears much resemblance to the “lipstick” sense chamber sources in early Pyrotronics detectors made in the USA. This one is brass and a bit wider than its Pyrotronics analogue. An internal axial thread positions a cup-shaped alpha particle shield around a band containing the active deposit, thereby regulating the amount of ionization produced by the source in the chamber and controlling the sensitivity of the detector. The sections below describe my analysis of the gamma and alpha radiations emitted by this source, and my conclusions about the plutonium’s age, activity, mode of production, and suitability for nuclear combat.



Plutonium isotopics by gamma spectrometry
High-resolution gamma spectroscopic measurements allow direct determination of the relative concentrations of Pu-238, Pu-239, Pu-240, and Pu-241 in a plutonium sample. In such measurements, Pu-242 is customarily inferred from heuristic correlations to the other isotopes; it can be directly measured only with costly and destructive mass spectrometry. Additionally, the ratio of daughter Am-241 to parent Pu-241 can be used to date plutonium. The basic methodology is discussed in good detail in Sampson, T. E., Plutonium Isotopic Composition by Gamma-Ray Spectroscopy (1986). I employed the multiple linear regression (MLR) formula of Sarkar, Shah, et al. (2014) to estimate Pu-242.
My gamma detector is a PGT n-type coaxial HPGe detector that lives in the guest bedroom of the home (the least radioactive room, as it should be), shielded with lead bricks and a graded inner shield of copper and tin sheet. One preparation that is almost essential with plutonium is selective attenuation of the 59-keV gamma radiation from Am-241, as discussed in Sampson’s article above. If you don’t do this, then the pileup and sum peaks caused by the intense Am-241 radiation will swamp the rest of the plutonium spectrum. To hold the “lipstick” source, I made an attenuator out of rolled cadmium sheet and endcaps stuffed inside of a piece of copper water pipe with copper endcaps. Such an arrangement works by strategically situating the K-edge energy of the absorber materials close to the energy of the offending radiation. In quantitative gamma spec measurements, another important point is to avoid getting the source too close to the detector. Otherwise, coincidences will distort the spectrum. With the right attenuation and geometry, all that remains is to gather a statistically-useful number of counts in the spectrum–in this case, about 42 hours of counting.



The gamma spectrum is shown, annotated, in the gallery below. It can be downloaded in ASCII format as an Excel spreadsheet here. (Note that there are no channel numbers or energy calibration in the ASCII format, so you will have to add them.) As can be seen, Am-241 and Pu-239 peaks are scattered throughout, while Pu-238, Pu-240, and Pu-241 are represented by a single good peak each in the 150-keV neighborhood. Am-241’s granddaughter Pa-233 is also in evidence, attesting to the unseen Np-237 daughter. U-237 is a product of the minor alpha decay branch of Pu-241, and it interferes with some lines in the Am-241 decay spectrum as both nuclides decay to Np-237. Those energies subject to interference cannot be used for quantitative analysis. Click any image for the larger original:




Calculating relative activities from the peaks in the spectrum involves the following:
- Measuring counts in each peak by peak-fitting algorithms. I use the free software Hypermet-PC 5.12 to do this. Its algorithms are old, but well-known and still widely used. Modern users will need to run it in DOSBox.
- Correcting measured counts by an efficiency function of energy. I fit this function in Hypermet-PC using a sealed Ra-226 source that can be placed in the same graded attenuator (and the same counting geometry) as the “lipstick” plutonium source.
- Calculating relative activities from efficiency-corrected counts using the tabulated yields per decay of each radiation. I used this website for my data.
- Estimating Pu-242 activity using a suitable model. My reference is here.
Once relative activities were established, I estimated total activities by comparing the gamma count rate on a Geiger counter between the KI-1 source and the ~60 microcurie Am-241 source from a Pyrotronics F-3/5A in the same counting geometry. The overwhelming majority of the gamma rays emitted by both sources are 59-keV photons from Am-241. These estimates are limited by the uncertainty surrounding the total activity of the Pyrotronics source. The relative activities are known to much higher precision. (I should note that the uncertainties given in the table relate to the relative measurements.) As the table below illustrates, the KI-1 source contains a total activity of about 700 microcuries today, most of which is the 14-year weak beta emitter Pu-241. The runner-up is 88-year alpha emitter Pu-238. On an activity basis, the other nuclides are lower in the lineup. The plutonium mass can be calculated, and it is about 1 mg.

The alpha spectrum
Alpha spectroscopy of plutonium is confounded by the fact that Pu-239 and Pu-240, and Pu-238 and Am-241, emit alpha particles with very similar energies. The general technique is also laborious, involving chemical preparation of samples in virtually all cases. Like Pyrotronics sources, there is some removable contamination on the KI-1 detector source. I wiped a tissue on the source surface, ashed it, dissolved the residues in nitric acid, and evaporated them onto a stainless steel disc to make the spectrum shown below using an Ortec solid-state detector. Despite this effort, it is not of great technical quality compared to what one could expect with a rigorous radiochemical technique. All that said, though: the spectrum confirms the expectation of two main alpha energy groups, the larger at 5.4-5.5 MeV (Pu-238+Am-241) and the smaller at 5.1-5.2 MeV (Pu-239+Pu-240).

Dating plutonium using the Am-241:Pu-241 ratio
The Am-241:Pu-241 atom ratio is a daughter-parent ratio, a clock that allows us to date the plutonium. More specifically, the method determines when Am was last chemically separated from the Pu, assuming that all the material in the source traveled together through the same process. (The assumption may not be very good if multiple batches of Pu were mixed.) A graphical solution of the coupled Bateman equations modeling Am and Pu ingrowth and decay is shown below. The sample age is the point on the horizontal axis where the solution intersects the measured value of Am-241:Pu-241, represented by the one-standard-deviation band between the red and blue lines. This plutonium appears to be 44.9 ± 0.4 years old, meaning it was probably processed in 1972.

Other dating ratios
Another member of the Pu-241 decay chain, Pa-233, can also be used for dating. In its ratio with Am-241, we get an estimate of 55.4 years; in its ratio with Pu-241, we get an estimate of 48.2 years. The Am-241:Pu-241 method above predicted 44.9 years. These three ages would be harmonized if there were a bit more Am-241 in the mix, specifically about 18% more, suggesting that some may have been removed in the earlier history of the sample. The removal may have coincided with initial fuel processing delayed appreciably after fuel discharge from the reactor, or it may have been undertaken some time after the initial processing. I am in favor of a view that americium was last chemically separated about four years after fuel discharge, the fuel itself being about 49 years out of the reactor (discharged in 1968), and that the separatory chemistry in the early 1970s was selective for Am and largely left ingrown Np-237 (parent of Pa-233) with the Pu. This hypothesis harmonizes all three age estimates.


Original plutonium composition
Armed with an age estimate and current activity ratios among all the Pu isotopes, the calculation of mass composition at the time of preparation is straightforward using tabulated values of the half lives (or decay constants) of the isotopes. Once again, there are assumptions in this calculation and in the conclusions derived from it. The most important is probably that the plutonium was “fresh” when it was processed (or, more specifically, that the time difference between when irradiation stopped and when processing occurred was small enough to be insignificant to the isotopics). Is that a good assumption? Because the half-life of Pu-241 is only 14 years, and because the logistics of nuclear fuel processing usually dictate several years of cool-down during which time the fuel is in storage, transit from the reactor, and standing in queue for processing, this number is perhaps most suspect–and we would expect its calculated value and that of the correlated Pu-242 estimate to err on the low side. Keeping this caveat in mind, here is the composition of the original KI-1 smoke detector plutonium as calculated from the Am-241:Pu-241 age:

What if the plutonium is actually four years older (1968) and was just processed in 1972, as the Pa-233 dating methods hint? Then, the composition looks like the table below. I believe this is more accurate:

Conclusions: Low-burnup, reactor-grade plutonium from 1970 is nothing to fear
With original Pu-240 concentration near 20%, the ~1 mg of plutonium used in this Soviet KI-1 smoke detector falls into the “reactor grade” classification rather than “weapon grade.” The classification convention distinguishes plutonium compositions on the basis of Pu-240 content because of this isotope’s high spontaneous-fission neutron yield and its consequences for pre-initiation in nuclear weapons. However, weapons made from reactor-grade plutonium are known to work. Their yield may not be statistically reliable or as high as could be expected with weapon-grade fissile material, but they are useful weapons nonetheless. The real barrier to would-be proliferants hoarding Soviet smoke detectors is the sheer number–millions!–of the motherfuckers they would in principle need to acquire through the typical nuclear smurfing networks. (The entire output of the Soviet smoke detector industry is unlikely to have involved more than one formula quantity of plutonium.)
Now that we can sleep easily on the nuclear holocaust issue, I’ll add a few more observations about this plutonium. Although reactor grade, its high fraction of Pu-239 and low fractions of Pu-241 and Pu-238 suggest moderately low burnup, probably not in excess of 5 GWd/t, in a reactor amenable to such light utilization (e.g. an isotope production reactor or online-refuelable type). The measured dates of production (1968) and last separation (1972) rule out VVER and RBMK power reactors as sources. Some of the RBMK’s graphite-moderated, low-enrichment-fueled predecessors designed for isotope production and co-located with processing plants (such as the ADE types) are likely origins. These reactors also turned out a weapon-grade stream as the USSR frantically raced for nuclear parity with the Yankee imperialists.