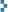
Uranium Chemistry
February 20, 2008 Uranium and its pure compounds are just not readily available to the amateur scientist, element collector, or student in 2008. So what is one to do? Make these materials oneself, of course. (At left is a quantity of home-baked yellowcake.)
Uranium and its pure compounds are just not readily available to the amateur scientist, element collector, or student in 2008. So what is one to do? Make these materials oneself, of course. (At left is a quantity of home-baked yellowcake.)
This is the inaugural post in what will become a short series, detailing how uranium and various pure compounds can be refined from the brute earth to serve personal needs. There are differences between what is done in industrial mining / milling operations and what can be realistically accomplished in a typical American domicile. There are also differences in the raw materials that could be obtained back in the good old days when our favorite applied inorganic chemistry texts were written (“Borrow a gallon of fuming nitric acid and some glycerin from your science-teacher…”), versus what can be obtained in the paranoid, restrictive world of today. Thus, my approach to uranium chemistry emphasizes practical techniques and materials that are available to today’s home-dweller. The foregoing discussion assumes a decent background in chemistry and mature attention to safety.

Uranium compounds that can be easily prepared at home are shown in this photo. In vials, left to right: uranyl oxide (UO3); uranyl peroxide (UO4·nH2O); triuranium octoxide, U3O8; sodium diuranate (Na2U2O7·6H2O); uranium tetrafluoride (UF4·2.5H2O); “sodium peruranate” in solution; uranyl chloride (UO2Cl2) in solution. In front is an electroplated layer of uranium dioxide (UO2). Click “more” below for content (I will upload it as time permits).
_______________________________________________________________
- Part I: Collecting uranium ore
- Part II: Crushing, milling; acid leaching
- Part III: Ammonia precipitation and carbonate extraction
- Part IV: Producing uranyl peroxide yellowcake
- Part V: Uranyl oxide (UO3); U3O8; uranyl salts
- Part VI: Producing uranium dioxide (UO2) by electrolysis
- Part VII: Producing uranium tetrafluoride (UF4)
- Part VIII: Odd stuff: “Sodium peruranate”
- Part IX: Producing uranium metal
_______________________________________________________________
Part I: Collecting uranium ore
 Uranium ore is easy to find on the abandoned uranium mine dumps and mine roads of the American Southwest. In fact, this is among the easiest kinds of rock to obtain by the bucketload, provided one has a car and is willing to spend a day or two in sunny Southeast Utah. You don’t need to know anything about geology, mining, or the privations of life in the backcountry as the “U-boomers” of the ’50s did (although some appreciation in these areas will enrich the experience). A high-clearance, 4WD vehicle is unnecessary; a Toyota Echo will get you to most dumps as surely as an H2 Hummer, and without branding you as an Earth-raping jackass. Summer weather is usually amenable to camping in the field. For those into luxury, there’s the Lazy Lizard Hostel in Moab.
Uranium ore is easy to find on the abandoned uranium mine dumps and mine roads of the American Southwest. In fact, this is among the easiest kinds of rock to obtain by the bucketload, provided one has a car and is willing to spend a day or two in sunny Southeast Utah. You don’t need to know anything about geology, mining, or the privations of life in the backcountry as the “U-boomers” of the ’50s did (although some appreciation in these areas will enrich the experience). A high-clearance, 4WD vehicle is unnecessary; a Toyota Echo will get you to most dumps as surely as an H2 Hummer, and without branding you as an Earth-raping jackass. Summer weather is usually amenable to camping in the field. For those into luxury, there’s the Lazy Lizard Hostel in Moab.
 The only critical equipment to this venture is a durable scintillation detector to indicate which rocks are hot (or where to dig to retrieve a hot rock). Unless you can see gamma rays, you just can’t look at the ground and discern a good piece of ore from a non-piece of ore. At left is an image of my homemade bismuth germanate scintillation detector, built in a section of water pipe and attached to a Ludlum 12 ratemeter. (Construction info here.) There are many little tricks to building and using scintillation detectors for this purpose, but a most important one is this: don’t hit the dumps with a new $1000 NaI(Tl) detector–you’ll kill it! I recommend plastic or BGO. I use high-resistance dynode chains, 10MΩ+ per dynode. This prolongs the life of the ratemeter batteries. Before departing on a trip, I set my high voltage so that the scintillometer pegs full scale on a nice piece of ore kept at home as a standard. In the field, I can roughly gauge the quality of a rock against my standard.
The only critical equipment to this venture is a durable scintillation detector to indicate which rocks are hot (or where to dig to retrieve a hot rock). Unless you can see gamma rays, you just can’t look at the ground and discern a good piece of ore from a non-piece of ore. At left is an image of my homemade bismuth germanate scintillation detector, built in a section of water pipe and attached to a Ludlum 12 ratemeter. (Construction info here.) There are many little tricks to building and using scintillation detectors for this purpose, but a most important one is this: don’t hit the dumps with a new $1000 NaI(Tl) detector–you’ll kill it! I recommend plastic or BGO. I use high-resistance dynode chains, 10MΩ+ per dynode. This prolongs the life of the ratemeter batteries. Before departing on a trip, I set my high voltage so that the scintillometer pegs full scale on a nice piece of ore kept at home as a standard. In the field, I can roughly gauge the quality of a rock against my standard.
Some other handy equipment should be common-sense to rockhounds:
- Bucket or backpack
- Geologist’s hammer (occasionally a mattock is useful)
- Topo maps, handheld GPS receiver
 Where are the best mines? Good question. I’m not sure I’ve figured that out (and should I really share my favorite spots)? But I can tell you a good mine to visit first. Here are instructions to the famous Mi Vida Mine, pictured left. From Moab, UT, head south about 30 mi. on US 191. Turn east at the junction of Steen’s Rd. (CR-114) and US 191. Follow Steen’s Rd. past CR-111, past the hairpin turn, and up a gently rising gulch until you see the big ore bins in the photo. This
Where are the best mines? Good question. I’m not sure I’ve figured that out (and should I really share my favorite spots)? But I can tell you a good mine to visit first. Here are instructions to the famous Mi Vida Mine, pictured left. From Moab, UT, head south about 30 mi. on US 191. Turn east at the junction of Steen’s Rd. (CR-114) and US 191. Follow Steen’s Rd. past CR-111, past the hairpin turn, and up a gently rising gulch until you see the big ore bins in the photo. This  is the Mi Vida Mine. It’s on BLM land, so feel free to fill your backpack with screaming hot rock. Focus on the road, and on the conspicuous dumps for the Standard Mine, slightly SW of the Mi Vida itself. If you continue east on Steen’s Rd. past the Mi Vida, the road gets rougher and you come out on the western wall of Big Indian Valley with impressive views of the La Sal Mountains to the north. This western escarpment is dotted with dozens of uranium mines. Get busy!
is the Mi Vida Mine. It’s on BLM land, so feel free to fill your backpack with screaming hot rock. Focus on the road, and on the conspicuous dumps for the Standard Mine, slightly SW of the Mi Vida itself. If you continue east on Steen’s Rd. past the Mi Vida, the road gets rougher and you come out on the western wall of Big Indian Valley with impressive views of the La Sal Mountains to the north. This western escarpment is dotted with dozens of uranium mines. Get busy!
_______________________________________________________________
Part II: Crushing, milling, and acid leaching
Big rocks must be turned into very small rocks in order to easily access the uranium in them with chemicals. Leaching solutions have a hard time getting inside big rocks. My method for turning big rocks into very small rocks is nothing fancy. I begin by placing big, brick-sized ore chunks on a very hard slab of other rock situated in the center of a shallow plastic bin out on the back porch, and I apply a crack hammer liberally, returning to attack any remnants larger than about the size of a pea. This is hard, manual labor. Anything passing through a 1/4″ chicken-wire mesh is done feeling the hammer, and anything passing a fine-screened kitchen sieve is considered leachable. The fraction passing the 1/4″ mesh but not the kitchen sieve is sent to be ball-milled.
 My ball mill was purchased on eBay from the seller Hobfir. This is his 15-lb model, entirely adequate for the quantity of rock I process and a very good deal. To give the mill drum better traction on the rollers, I apply duct tape around the circumference of the drum and a few drops of honey on the rollers. Works wonders at keeping that puppy rollin’. The milling media is a mixture of 1/2″ and 3/4″ chrome steel balls purchased from McMaster-Carr. I tend to put in about 5 lb of balls to 5 lb of crushed ore. A day’s worth of milling turns most of the charge into powder that passes my kitchen sieve.
My ball mill was purchased on eBay from the seller Hobfir. This is his 15-lb model, entirely adequate for the quantity of rock I process and a very good deal. To give the mill drum better traction on the rollers, I apply duct tape around the circumference of the drum and a few drops of honey on the rollers. Works wonders at keeping that puppy rollin’. The milling media is a mixture of 1/2″ and 3/4″ chrome steel balls purchased from McMaster-Carr. I tend to put in about 5 lb of balls to 5 lb of crushed ore. A day’s worth of milling turns most of the charge into powder that passes my kitchen sieve.
 Leaching uranium can be done with acids, or with carbonates. In industry, the choice depends on ore chemistry. Acid is uneconomical if the ore has lots of carbonates in it, but acid is more aggressive. Bob Lazar of UnitedNuclear.com describes a carbonate leach process here. I like an acid process with this Utah ore—-acid reacts very quickly and completely, even though it is not at all selective for uranium. My acid leachate goes on to feed a secondary carbonate leach, so in a sense I am only using the acid to break down / pulverize the ore further than the ball mill. The acid used here is “muriatic acid” (20° Baume HCl solution sold at Lowes and Home Depot), straight out of the jug. The ore is first wet with water to make a fluid slurry in the bottom of a 5-gallon bucket, and then HCl is added. A large quantity of CO2 is evolved as the acid reacts with calcite and dolomite in the sandstone. I periodically stir the leaching bucket to resuspend the solids, and I add HCl to keep pH < 0.
Leaching uranium can be done with acids, or with carbonates. In industry, the choice depends on ore chemistry. Acid is uneconomical if the ore has lots of carbonates in it, but acid is more aggressive. Bob Lazar of UnitedNuclear.com describes a carbonate leach process here. I like an acid process with this Utah ore—-acid reacts very quickly and completely, even though it is not at all selective for uranium. My acid leachate goes on to feed a secondary carbonate leach, so in a sense I am only using the acid to break down / pulverize the ore further than the ball mill. The acid used here is “muriatic acid” (20° Baume HCl solution sold at Lowes and Home Depot), straight out of the jug. The ore is first wet with water to make a fluid slurry in the bottom of a 5-gallon bucket, and then HCl is added. A large quantity of CO2 is evolved as the acid reacts with calcite and dolomite in the sandstone. I periodically stir the leaching bucket to resuspend the solids, and I add HCl to keep pH < 0.
Acids will only extract significant uranium if it is in the U(VI) oxidation state. Therefore, an oxidizer is added to the leach pail to help get the U(IV) in the mixed-state pitchblende into the U(VI) state. Being cheap, I prefer chlorine bleach. (You should be getting a picture of why it is important that this process be done outside!) I add bleach, stir, and repeat, dodging the cloud of chlorine gas wafting out of the bucket. The leachate is a dark forest-green color before bleach is added, but after we’re “done,” it becomes a lighter apple-green hue owing to the oxidation of Fe(II) to Fe(III). The iron is actually a catalyst in this leaching process: when in the Fe(III) state, it will oxidize U(IV). Bleach is alkaline, so typically I have to adjust the pH again after an addition of bleach. Other oxidants will do the job at considerably higher cost: KClO3, KMnO4, MnO2, and H2O2.
 Finished leachate is siphoned off after several days of sitting / stirring. It is apple-green, and very turbid despite efforts to give it several days to settle out. I gravity-filter it using coffee filters, coffee strainers, and plastic funnels. Then I vacuum-filter it through fine qualitative filter paper. The result is shown at left.
Finished leachate is siphoned off after several days of sitting / stirring. It is apple-green, and very turbid despite efforts to give it several days to settle out. I gravity-filter it using coffee filters, coffee strainers, and plastic funnels. Then I vacuum-filter it through fine qualitative filter paper. The result is shown at left.
Summary: The acid leaching process described above comprises the following reactions:
Oxidation and dissolution of U(IV): UO2(s) + Cl2(aq) → (UO2)2+(aq) + 2Cl–(aq)
Dissolution of U(VI): UO3(s) + 2H+(aq) → (UO2)2+(aq) + H2O(l)
_______________________________________________________________
Part III: Ammonia precipitation and carbonate extraction

Carbonate solutions are good solvents for U(VI), holding uranium in solution when most other elements that precipitate with ammonia remain insoluble. At left is pictured the product of this step: a yellow and very fluorescent solution containing uranylcarbonate anions, e.g. UO2(CO3)3-4. Before getting to this point, however, the acid leachate from Part II is reacted with ammonia to precipitate most of the transition metals and uranium, while leaving the alkali metals and certain other contaminants dissolved. This step requires the following chemicals:
- Household ammonia (I insist on Ace Hardware Janitorial Strength Ammonia for any kind of chemistry project. It comes in a gallon jug, it’s very strong, and it’s the only ammonia sold in my area that I know to be free of surfactants.)
- Sodium carbonate, Na(CO3)2. (The anhydrous material can be found in the pool supply section of Lowe’s or Home Depot, or the decahydrate Na(CO3)2·10H2O can be bought at a grocery as Arm and Hammer Super Washing Soda. I prefer the latter for reasons of cost and availability, and my mass quantities given here pertain to the use of washing soda.)
- Sodium bicarbonate
We start with green HCl leachate made in Part II. Enough household ammonia is stirred into the leachate to raise the pH to 10. A voluminous precipitate forms. At pH = 5 the solids are yellow, reflecting the precipitation of uranium as diuranate (U2O72-). As more ammonia is added, the solids become a dark olive-green color. Some people might argue that I should stop adding NH3 at pH = 5-7 and achieve some selectivity for the uranium. That might be a good idea; I haven’t tried it. My reason for adding to pH = 10 is to (A) ensure complete precipitation of the uranium and (B) have excess NH3 to complex Zn, Cu, Ni, Ag, and Cd. Anyway, an airtight cover is placed on the container and the precipitate allowed to settle. After settling, the clear, purplish supernatant solution containing the ammonia complexes is decanted and the precipitate is washed and settled 3-5 times in boiling tapwater to remove ammonia and soluble salts. Drying the precipitate is unnecessary, but if done, a yellowish-brown powder is obtained, as shown at left.
(B) have excess NH3 to complex Zn, Cu, Ni, Ag, and Cd. Anyway, an airtight cover is placed on the container and the precipitate allowed to settle. After settling, the clear, purplish supernatant solution containing the ammonia complexes is decanted and the precipitate is washed and settled 3-5 times in boiling tapwater to remove ammonia and soluble salts. Drying the precipitate is unnecessary, but if done, a yellowish-brown powder is obtained, as shown at left.
Carbonate leaching of the ammonia precipitate will be used to separate uranium and a very few other metals (vanadium, molybdenum) from everything else (probably mostly iron). I could simply leach the crushed ore with carbonate, but in my experience, carbonate is slow to attack Utah ore. The acid leach / ammonia precipitation essentially are used here to efficiently produce a more concentrated, finely-divided, and oxidized feed for the carbonate leach. My lixiviant recipe is as follows: in a five-gallon pail, dissolve 2 kg of washing soda in 13 l of hot tap water. This solution is too alkaline to dissolve uranium, so must be buffered at pH 10.5 with baking soda. 2-3 boxes seem to be required. Atmospheric CO2 is also part of this equilibrium.
 The leaching itself is fun: I made a leaching system comprising a small microwave oven, a length of PE tubing, a pail, and a submersible aquarium pump, see photo left. Two small holes are drilled in the wall of the oven cavity (they are too small to radiate microwaves). A length of 1/4″ PE tubing is loosely coiled inside the oven and brought out through the holes. The submersible pump sits in the leach pail and drives solution through the coil and back into the pail. Discharge temperature of this coil is close to 80 °C after the system has been running awhile. Obviously, some care went into determining the right length of tubing; you don’t ever want to boil its contents! The pail is charged about half full with
The leaching itself is fun: I made a leaching system comprising a small microwave oven, a length of PE tubing, a pail, and a submersible aquarium pump, see photo left. Two small holes are drilled in the wall of the oven cavity (they are too small to radiate microwaves). A length of 1/4″ PE tubing is loosely coiled inside the oven and brought out through the holes. The submersible pump sits in the leach pail and drives solution through the coil and back into the pail. Discharge temperature of this coil is close to 80 °C after the system has been running awhile. Obviously, some care went into determining the right length of tubing; you don’t ever want to boil its contents! The pail is charged about half full with  carbonate solution, and the ammonia precipitate produced by neutralization of 2 l of acid leachate is added. After starting the pump and oven, a uranium yellow color is evident in the carbonate solution almost immediately. I typically operate this system for a few hours in the evening over 2-3 days. The consumption of CO32- by uranium drives the pH up noticably, so baking soda must be occasionally added to keep the pH around 10.5. Pregnant leachate is extracted when needed by directing the heater coil discharge into a funnel with a coffee filter in it. A receptacle under the funnel collects the clear yellow solution shown at the top. I subsequently vacuum-filter
carbonate solution, and the ammonia precipitate produced by neutralization of 2 l of acid leachate is added. After starting the pump and oven, a uranium yellow color is evident in the carbonate solution almost immediately. I typically operate this system for a few hours in the evening over 2-3 days. The consumption of CO32- by uranium drives the pH up noticably, so baking soda must be occasionally added to keep the pH around 10.5. Pregnant leachate is extracted when needed by directing the heater coil discharge into a funnel with a coffee filter in it. A receptacle under the funnel collects the clear yellow solution shown at the top. I subsequently vacuum-filter  the solution through fine qualitative filter paper to remove all traces of turbidity. Oh, and I should mention the fluorescence! The uranyl ion glows a brilliant green color under long- and short-wave UV.
the solution through fine qualitative filter paper to remove all traces of turbidity. Oh, and I should mention the fluorescence! The uranyl ion glows a brilliant green color under long- and short-wave UV.
_______________________________________________________________
Part IV: Producing uranyl peroxide yellowcake
First of all, “yellowcake” is a not a specific chemical compound. In industry, the word simply refers to uranium mill concentrates, regardless of their chemistry. Yellowcakes may consist of alkali or ammonium uranates or diuranates and / or uranium oxides. In most processes today, the mill concentrate is U3O8, which is not even yellow! Here we precipitate a yellowcake of uranyl peroxide (UO4·nH2O) that is indeed a very satisfying shade of yellow. More importantly, this precipitation is highly selective for uranium. Uranium peroxide is one of few inorganic peroxides that are insoluble in water. In fact, the only known naturally-occurring peroxide mineral is studtite — uranyl peroxide.
The uranylcarbonate solution from Part III is divided into two portions, one large (about 700 ml / l of original solution) and one small (about 300 ml / l). The large portion is neutralized with muriatic acid to pH = 3, accompanied by much effervescence of CO2, and then boiled to remove dissolved CO2. Some precipitate may fall out during the neutralization process and this should be kept in the liquid. The small portion of solution is set aside for later.
Hydrogen peroxide in excess is added to the de-carbonated large portion of the solution from above while it is still boiling hot. The hotter the solution, the more manageable the precipitate. My preferred source of peroxide is Baquacil Ultra, 27% H2O2 sold locally by the gallon as a swimming pool oxidizer. I add about 20 ml Baquacil / l solution. (Note, 27% H2O2 is feisty stuff, quite unlike 3% drugstore H2O2. It will flense the living skin right off a dude by way of large white burns.) Two results obtain: a fine light-yellow precipitate quickly falls out of solution, and the pH falls dramatically. As the summary at bottom illustrates, this is due to acid formation that accompanies uranyl peroxide formation.
Here’s where the small portion of uranylcarbonate solution comes in. The remaining uranylcarbonate solution is used to neutralize acid formed by peroxide addition and return the pH to the range of 3-4. Stir quickly during this addition, which like the above step should be completed while the main solution is still almost boiling hot. I typically reach pH = 3.5 with uranylcarbonate to spare. I simply return the excess to the container whence it came and save it for another batch. A brief reddish or orange coloration may be noted upon addition of the carbonate solution—-see Part VIII below for the explanation.
Finishing up: Allow the precipitate to continue forming and settling for several hours in a covered container. Decant the liquid and wash several times with boiled distilled water. Dry in a glass casserole dish via microwave oven.
Summary: The impure uranylcarbonate made in Part III is neutralized with acid to form a uranyl solution, and an insoluble uranium peroxide is selectively precipitated from the uranyl solution to free the uranium from impurities. It should be noted that the stoichiometry of the uranylcarbonate anion and the uranyl peroxide water adduct depend on certain uncontrolled variables, so the below balanced reactions are only generally representative.
Neutralization: 6H+(aq) + UO2(CO3)34-(aq) → (UO2)2+(aq) + 3CO2(g) + 3H2O(l)
Precipitation: (UO2)2+(aq) + H2O2(l) → UO4·2H2O(s) + 2H+(aq) + 2H2O(l)
_______________________________________________________________
Part V: Uranyl oxide (UO3); U3O8; uranyl salts
 Uranium as the peroxide from Step IV can be purified further by converting it to UO3 or U3O8. Fiesta-orange UO3 is the final product of my purification routine, and the gateway to other uranium compounds. It is produced by heating dry UO4·nH2O in an open-air container at about 300 °C. During the process of heating, volatile contaminants (water, oxygen, ammonia, halogens, hydrogen peroxide) are driven off. During preparation, keep stirring the yellow peroxide as it turns orange. If the temperature is managed properly, the product will be a clean orange color. If allowed to overheat, U3O8 begins forming and the orange color fades to darker shades of brown-green. What I like about UO3 as an endpoint is that this oxide is readily acid-soluble, has definite stoichiometry, and does not require a lot of heat to form. I store my “stock” of uranium as UO3.
Uranium as the peroxide from Step IV can be purified further by converting it to UO3 or U3O8. Fiesta-orange UO3 is the final product of my purification routine, and the gateway to other uranium compounds. It is produced by heating dry UO4·nH2O in an open-air container at about 300 °C. During the process of heating, volatile contaminants (water, oxygen, ammonia, halogens, hydrogen peroxide) are driven off. During preparation, keep stirring the yellow peroxide as it turns orange. If the temperature is managed properly, the product will be a clean orange color. If allowed to overheat, U3O8 begins forming and the orange color fades to darker shades of brown-green. What I like about UO3 as an endpoint is that this oxide is readily acid-soluble, has definite stoichiometry, and does not require a lot of heat to form. I store my “stock” of uranium as UO3.
If U3O8 is desired, simply heat orange UO3 until it is uniformly dark brownish-green. This takes a lot of heat, and produces a mixed-valence product of variable stoichiometry that has some difficulty dissolving in acid. One neat trick in the preparation of U3O8 is to exploit its large dielectric loss tangent by finishing the reaction from UO3 in a microwave oven. Fill a large alumina crucible about half full with with a “seed layer” of finished U3O8 and top off the crucible with fresh UO3. The lossy U3O8 becomes orange-hot and the heat finishes the conversion of the fresh UO3.
_______________________________________________________________
Part VI: Producing uranium dioxide (UO2) by electrolysis
 Thin layers of UO2 can be electroplated onto metal cathodes in a uranyl salt bath. The technique is not efficient from an electrochemical standpoint (the vast majority of input energy goes toward production of H2 and O2 and heat), but potentially very handy for production of UO2 semiconductor components or fabrication of experimental fission chambers.
Thin layers of UO2 can be electroplated onto metal cathodes in a uranyl salt bath. The technique is not efficient from an electrochemical standpoint (the vast majority of input energy goes toward production of H2 and O2 and heat), but potentially very handy for production of UO2 semiconductor components or fabrication of experimental fission chambers.
Supplies needed include the following:
- UO3 (see Part V)
- Dilute sulfuric acid (e.g. battery acid from Pep Boys or Checkers)
- Ammonia (Ace Hardware Janitorial Strength)
- An accurate pH meter
- Cathode(s) to be plated; I used copper sheet and box cutter blades with success.
- An anode that will not get oxidized. I recommend the platinum-plated titanium anode from Jumpin’ Jack Flash Pyro Supplies. It has many uses!
- 15 V, 10 A DC power supply
- A plastic or glass container
In my experiment I used a plastic ~300 ml food-storage container as the electrolysis cell. In the future I plan on using glass because of the ohmic heating of the electrolyte. To make the electrolyte, I dissolved 6 g UO3 in battery acid and then partially neutralized with ammonia to pH = 2.0. The resulting solution was placed into the electrolysis cell and diluted to ~300 ml with a 0.2 M (NH4)2SO4 solution made earlier from acid and ammonia. Finally, the pH was adjusted to 2.5 with a few drops of acid. Time to turn on the juice!
 The electrolysis setup is assembled by arranging the anode on the vertical axis of the cylindrical cell and clipping the cathode to the wall. See photo at left. I have had best results in my experiments when the cathode presents no more than about 15 cm2 of area in the direction of the anode. (The “back” of the cathode will be minimally plated except around the periphery.) The power supply is turned on and electrolysis begins with heavy evolution of gas at both electrodes. During electrolysis, the solution becomes hot and current may rise or fall. I move the cell to bring the anode closer to or farther from the cathode to help hold current to about 10 A. The finished plating is an adherent, velvety black layer. I plated my cathodes for about 5 min. apiece; longer plating tends to make a less adherent layer that can crack off, although more UO2 is present. Between platings, electrolyte level can be restored with distilled water.
The electrolysis setup is assembled by arranging the anode on the vertical axis of the cylindrical cell and clipping the cathode to the wall. See photo at left. I have had best results in my experiments when the cathode presents no more than about 15 cm2 of area in the direction of the anode. (The “back” of the cathode will be minimally plated except around the periphery.) The power supply is turned on and electrolysis begins with heavy evolution of gas at both electrodes. During electrolysis, the solution becomes hot and current may rise or fall. I move the cell to bring the anode closer to or farther from the cathode to help hold current to about 10 A. The finished plating is an adherent, velvety black layer. I plated my cathodes for about 5 min. apiece; longer plating tends to make a less adherent layer that can crack off, although more UO2 is present. Between platings, electrolyte level can be restored with distilled water.
 Plated cathodes read about 2000-3000 CPM on a pancake GM tube, indicating that the black deposit is indeed the intended UO2. If the cathodes are dirtied with precipitated uranyl salts, they can be washed by a quick dunk in dilute HCl and the UO2 layer will be unaffected. I have successfully used a UO2-plated copper strip in a “crystal radio” with the diode junction formed between it and a tuft of steel wool. The radio works, but poorly. I need to continue experimenting with junction materials and bias to better understand the semiconductor properties of UO2.
Plated cathodes read about 2000-3000 CPM on a pancake GM tube, indicating that the black deposit is indeed the intended UO2. If the cathodes are dirtied with precipitated uranyl salts, they can be washed by a quick dunk in dilute HCl and the UO2 layer will be unaffected. I have successfully used a UO2-plated copper strip in a “crystal radio” with the diode junction formed between it and a tuft of steel wool. The radio works, but poorly. I need to continue experimenting with junction materials and bias to better understand the semiconductor properties of UO2.
Summary: U(VI) is reduced to U(IV) at the cathode of an electrochemical cell. The intermediates and byproducts in this process seem to not all be well-known.
This procedure is adapted from a paper by Maya and Gonzalez entitled “Electrodeposition of uranium dioxide films,” J. Radioanalytical and Nuclear Chemistry, 261(3) 605-607 (2004). I link to the Springer full text reluctantly—-Springer is a major Google-spammer and does not deserve your money.
_______________________________________________________________
Part VII: Producing uranium tetrafluoride
 Uranium tetrafluoride (UF4), known as “green salt” in the uranium processing business, is an important intermediate in the production of both uranium metal and the uranium hexafluoride needed for gaseous diffusion enrichment. It is insoluble in water and one of the most well-behaved U(IV) compounds. The procedures described here produce a hemipentahydrate adduct, UF4·2.5H2O.
Uranium tetrafluoride (UF4), known as “green salt” in the uranium processing business, is an important intermediate in the production of both uranium metal and the uranium hexafluoride needed for gaseous diffusion enrichment. It is insoluble in water and one of the most well-behaved U(IV) compounds. The procedures described here produce a hemipentahydrate adduct, UF4·2.5H2O.
 This procedure starts with a trip to the grocery store to pick up a bottle of Whink Rust Stain Remover (“as advertised on Dr. Laura”) and a bottle of plain 1000 mg Vitamin C tablets. I caution the reader (and Dr. Laura’s braindead Republican audience) that Whink is hardly a benign household chemical—-it is 2-3M HF, and in this procedure, a solution containing it gets heated to boiling! Plan on being careful and do not breathe any fumes. In this process, I reduce uranium from U(VI) to U(IV) in solution with ascorbic acid and a Cu(II) catalyst, and simultaneously precipitate the U(IV) so formed with HF. Cl– ions are necessary also, so the solvent here is dilute HCl prepared from muriatic acid. In the image at left, I have the reactants laid out. They are as follows:
This procedure starts with a trip to the grocery store to pick up a bottle of Whink Rust Stain Remover (“as advertised on Dr. Laura”) and a bottle of plain 1000 mg Vitamin C tablets. I caution the reader (and Dr. Laura’s braindead Republican audience) that Whink is hardly a benign household chemical—-it is 2-3M HF, and in this procedure, a solution containing it gets heated to boiling! Plan on being careful and do not breathe any fumes. In this process, I reduce uranium from U(VI) to U(IV) in solution with ascorbic acid and a Cu(II) catalyst, and simultaneously precipitate the U(IV) so formed with HF. Cl– ions are necessary also, so the solvent here is dilute HCl prepared from muriatic acid. In the image at left, I have the reactants laid out. They are as follows:
- 2 g CuSO4·5H2O (Roebic K-77 root killer from Home Depot)
- 10.2 g UO3
- 100 mL Whink
- 20 mL muriatic acid
- Boiling distilled water
- (7) 1000 mg Vitamin C tablets
Dissolve the UO3 into the HCl and ~50 mL boiling water in an Erlenmeyer flask or similar container to form a yellow uranyl solution. Add the CuSO4 and Whink, add a glass chip (to help boiling later on) and set aside. Crush the Vitamin C tablets. In another container, stir the Vitamin C powder with ~50 mL boiling distilled water to dissolve as much of the material as possible. This sludgy solution will have to be filtered, probably several times. Vacuum filtration is almost required due to the gloppy binder mixed with the vitamin. If you can find pure Vitamin C powder at your stores, this is certainly preferable to tablets. I was not so lucky. With the ascorbic acid solution at the ready, heat the uranyl solution to boiling in a microwave oven. Once boiling, retrieve from the oven and stir in a few mL of the ascorbic acid. The uranyl solution immediately turns a bluish-green hue, with a flash of yellowish right where the ascorbic acid enters, and a precipitate forms. Return to microwave, boil, add ascorbic acid, and repeat until the latter is consumed. At this point the flask contains Cu+ and Cu++ in solution and a fine UF4 precipitate. Let flask sit an hour to cool down and settle precipitate (picture at left). Decant the supernatant solution. Wash and settle the precipitate twice in boiling water. Dry in microwave. The product should appear as at the top of this section. My yield was 12.6 g, nearly quantitative.
acid. The uranyl solution immediately turns a bluish-green hue, with a flash of yellowish right where the ascorbic acid enters, and a precipitate forms. Return to microwave, boil, add ascorbic acid, and repeat until the latter is consumed. At this point the flask contains Cu+ and Cu++ in solution and a fine UF4 precipitate. Let flask sit an hour to cool down and settle precipitate (picture at left). Decant the supernatant solution. Wash and settle the precipitate twice in boiling water. Dry in microwave. The product should appear as at the top of this section. My yield was 12.6 g, nearly quantitative.
My inspiration for this procedure comes from US Patent 3023078 (1962), “Production of Uranium Tetrafluoride,” by Robert J. Allen and Henry G. Petrow and assigned to the USAEC. They prefer a reduction with sulfur dioxide rather than Vitamin C, but for the kitchen chemist the latter is far easier to use. I also experimented with an H2S reduction of a mildly acidic uranyl chloride solution and had success, but at the expense of great effort to build the closed gas-flow system and make aluminum sulfide needed to feed the gas generator. H2S is very nasty stuff.
Summary: U(VI) is indirectly reduced to U(IV) by ascorbic acid (which is oxidized to dehydroascorbic acid), and precipitated with F–. Cu and Cl– are catalysts and bystanders and are omitted from the accounting below, though very necessary for success.
Reduction: (UO2)2+(aq) + C6H8O6(aq) + 2H+(aq) → U4+(aq) + C6H6O6(aq) + 2H2O(l)
Precipitation: U4+(aq) + 4HF(aq) + 2.5H2O(l) → UF4·2.5H2O(s) + 4H+(aq)
_______________________________________________________________
Part VIII: Odd stuff: “Sodium peruranate.”

The reaction of uranyl peroxide and sodium carbonate forms a blood-red solution that when more dilute appears dark orange. What’s particularly odd about the compound is that when it is generated in a 70% isopropanol (rubbing alcohol) solution, the solution separates into two transparent liquid layers–one blood-red, the other golden. You can shake the mixture all you want, but like oil and vinegar, the liquids separate again with the uranium “blood” on the bottom. Both liquids are soluble in excess water. Neither sodium carbonate nor uranyl peroxide by themselves form such an oil with isopropanol. Clean sodium hydroxide forms a yellow solution; the carbonate is evidently necessary for the red liquid precipitate to form.
In 1903, a report by J. Aloy entitled “A New Class of Peruranates” appearing in The Chemical News mentions the following:
“A solution of nitrate of uranium is treated with peroxide of hydrogen in sufficient quantity to precipitate the whole of the metal; wash by decantation and add some solid alkaline carbonate. In this manner we obtain a red solution which, when treated with ordinary alcohol, gives a red oil […]”
Aloy goes on to describe a method for precipitating a dark red unstable solid “peruranate” with methanol. He assigned it a formula of UO5Na2·5H2O based on quantitative analysis.
_______________________________________________________________
Part IX: Producing Uranium Metal
First off, I should point out that certain aspects of this high-temperature metallothermic reduction are not exactly “kitchen chemistry.” I am reducing anhydrous UF4 with finely-divided calcium metal in a special MgO crucible. The bill of materials is as follows:
- UF4·2.5H2O (from Part VII)
- ~25 g granular calcium metal, 8 mesh (purchased on eBay, $55 for 500 g)
- A 100-ml MgO straight-wall crucible, from Ozark Technical Ceramics ($35)
- 1 kg MgO powder, from Ozark Technical Ceramics ($10)
- An empty paint can
- Sand
- A short piece of Mg ribbon
- ~5 g crushed iodine
- Ice-cold white vinegar
- Acetone
- A vacuum pump
- A vacuum chamber rated to ~400 °C
That’s quite a list of stuff, some of it rather esoteric and costly. Additionally, this reaction absolutely must be done outdoors, away from combustibles and other people. Before getting too excited and pulling out your wallet, be sure to read the results below. Although this reaction does produce uranium metal, the scale is evidently too small to neatly separate the metal from the slag mixture in a “derby”.
 The UF4 used in a metallothermic reduction must be anhydrous. If it’s not, the water of hydration may consume the fuel metal, and steam may cause the reaction to erupt or splatter dangerously. UF4·2.5H2O cannot be dehydrated in air (it will react with oxygen to give uranium oxides), so a vacuum system is called for.
The UF4 used in a metallothermic reduction must be anhydrous. If it’s not, the water of hydration may consume the fuel metal, and steam may cause the reaction to erupt or splatter dangerously. UF4·2.5H2O cannot be dehydrated in air (it will react with oxygen to give uranium oxides), so a vacuum system is called for.  I use a 2.75″ ConFlat nipple linked by a long run of 1/4″ copper tubing to a two-stage mechanical vacuum pump. A quantity of UF4·2.5H2O is sealed into the chamber, the pump is started, and the chamber is directly heated on “high” on a gas stovetop. Progress may be checked via a simple electrodeless discharge tube mounted at the pump inlet and excited by a “leak tester” Tesla coil. When the grey-purple water discharge disappears, she’s done. My job took almost three hours.
I use a 2.75″ ConFlat nipple linked by a long run of 1/4″ copper tubing to a two-stage mechanical vacuum pump. A quantity of UF4·2.5H2O is sealed into the chamber, the pump is started, and the chamber is directly heated on “high” on a gas stovetop. Progress may be checked via a simple electrodeless discharge tube mounted at the pump inlet and excited by a “leak tester” Tesla coil. When the grey-purple water discharge disappears, she’s done. My job took almost three hours.
 Mixing and igniting the charge: The anhydrous UF4 was cooled, removed from the vacuum pump, and mixed with granular calcium. I prepared 67 g of UF4, and in a tightly-capped plastic bottle I shook this with 21 g calcium (about 25% stoichiometric excess). The mixture was then packed into the 100-ml MgO crucible, filling it about 2/3 full. Packability is
Mixing and igniting the charge: The anhydrous UF4 was cooled, removed from the vacuum pump, and mixed with granular calcium. I prepared 67 g of UF4, and in a tightly-capped plastic bottle I shook this with 21 g calcium (about 25% stoichiometric excess). The mixture was then packed into the 100-ml MgO crucible, filling it about 2/3 full. Packability is  very poor with this fluffy powder. A short piece of Mg ribbon was inserted into the center of the charge. Finally, a layer composed of about 5 g crushed iodine mixed with a pinch more of Ca metal was sprinkled atop the charge to help initiate the reaction, and provide CaI2 to lower the melting point of the slag. The crucible was surrounded with dry MgO powder in a paint can; this in turn was placed in a container of sand. Ignition is straightforward: light the Mg ribbon. Avoiding the smoke is probably a very good idea! Click here to watch the video (AVI format, 10.0 MB).
very poor with this fluffy powder. A short piece of Mg ribbon was inserted into the center of the charge. Finally, a layer composed of about 5 g crushed iodine mixed with a pinch more of Ca metal was sprinkled atop the charge to help initiate the reaction, and provide CaI2 to lower the melting point of the slag. The crucible was surrounded with dry MgO powder in a paint can; this in turn was placed in a container of sand. Ignition is straightforward: light the Mg ribbon. Avoiding the smoke is probably a very good idea! Click here to watch the video (AVI format, 10.0 MB).
 After sitting about three hours, the crucible was cool enough to recover the slag. In this experiment, the nature of the slag varied with depth in the crucible: at the top was some unconsolidated granular material that was minimally radioactive, followed by dense, brownish-black, consolidated slag at the bottom that was quite radioactive. (The crucible itself was destroyed by the reaction, breaking into several pieces). I retained the black bottom-slag, and found it to have the following properties:
After sitting about three hours, the crucible was cool enough to recover the slag. In this experiment, the nature of the slag varied with depth in the crucible: at the top was some unconsolidated granular material that was minimally radioactive, followed by dense, brownish-black, consolidated slag at the bottom that was quite radioactive. (The crucible itself was destroyed by the reaction, breaking into several pieces). I retained the black bottom-slag, and found it to have the following properties:
- Measurable (but poor) electrical conductivity
- Strongly radioactive (a couple chunks read 150 kCPM on a pancake GM tube)
- Flammable (if ignited, black powder will spall off and orange clinkers will “worm” through the mass)
- Evolves H2 gas from dilute HCl (photo below)
- Does not evolve noticeable gas from vinegar


I crushed the slag and attempted to wash it in ice-cold vinegar (a technique for dissolving calcium oxide / nitride / iodide, hopefully breaking up the slag and allowing isolation of the entrapped uranium metal). The slag as a whole was very resistant to vinegar, but some calcium salts evidently did dissolve because mixing acetone with the decanted vinegar resulted in a characteristic “Sterno” precipitate. Of course, vinegar will not dissolve the main expected byproduct, CaF2.
Conclusions and summary:
UF4(s) + 2Ca(s) → U(l) + 2CaF2(l)
My metallothermic reduction in a 100-ml crucible did indeed produce some uranium metal. I can infer as much from the slag’s flammability, electrical conductivity, radioactivity, and displacement of H2 gas from HCl. However, the uranium is bound up in a hard and chemically intractable CaF2 slag, along with other impurities that probably include uranium oxides. When these reactions are done on a large scale, they apparently work quite well, generating and storing enough heat to separate out molten uranium metal as a derby that settles to the bottom of the vessel. If one can’t isolate the metal in relatively pure form, then there is really no practical point to the exercise! In the future, I will probably explore the electrolytic synthesis of the metal described by Shiokawa et al. (J. Alloys and Compounds 255 (1997) p. 98-101). This ain’t exactly kitchen chemistry, either: it involves amalgamation of the product on a mercury cathode, which then must be distilled to isolate the U.

Carl, An excellent presentation. The reduction video is most impressive! Colored flames, sparkles, glowing slag — what more could you want?
I hope you have a discrete source for the I2 that you mentioned. A few years back I bought 100 gm of crystalline I2 off eBay for use in plasma globe experiments, and about a year later, I had a visit from two nice DEA agents with guns inquiring what I intended to do with it. Turns out that I2 is an ingredient in making crystal meth, and is on a list of proscribed substances. See:
http://www.deadiversion.usdoj.gov/21cfr/cfr/1310/1310_02.htm#a
Neat stuff!
Dave
Extreme detail and in what I would think is somewhat obscure subject. Great work.
Dave, Mark,
Thanks for your comments.
Dave, I obtained ~100g of iodine back in my high-school days and it has lasted, despite a variety of uses. The DEA’s attempt to crack down on drugs by restricting access to general-purpose materials that have use as precursors is becoming laughable; it’s probably symptomatic of the frustration that comes from being unable to control the drugs themselves. Oh well…amateur chemistry will go on, DEA or no DEA.
Hello Mr. Willis,
In the step when the uranium peroxide is precipitated out, can 6 %, pharmacy, hydrogen peroxide be used?
Thanks,
Thanks Carl for an informative FAQ – much appreciated and one for the archieve.
Carl,
This is a great website! You are,as always a true inspiration…now in your own turn. I see you believe in “pass it on”.
I feel with delight that a “torch is passed”…. Past into the best of hands with the best of hearts.
Richard
Hi, I did something similar a few years ago.
Your presentation and understanding is better
than mine, but check it out.
http://www.geocities.com/norm_alara/
Nice work, Carl!! Very impressive.
I probably shouldn’t comment at all before reading at least some of the text, but my instinct informs me that this is something quite out of the ordinary—it’s something to do with the imagery, I think.
Very mysterious?
Though HE dets are what turn me on nuclear (or nukillear as George W might say) detonations have a Majestic, terrible beauty all their own, and, yes, I know this isn’t a vey apt comment but like this page, somewhat out of the ordinary, possibly.
P
An extremely interesting presentation Carl, very well documented too.
Have you considered trying to produce uranyl nitrate or uranyl acetate? They’re both interesting compounds, delightfully fluorescent, thanks to the ion as you’ve already noted, and they’re of some utility as soluble uranium reagents for electron microscopy stains and things.
ion as you’ve already noted, and they’re of some utility as soluble uranium reagents for electron microscopy stains and things.
Some people out there in the community might be alarmed by the chemistry that you’ve demonstrated in your home – I’m not alarmed at all. What alarms me is that they actually sell 3M hydrofluoric acid to any idiot who walks into a store, in your country 😮 😉
[…] lots of interesting stuff. (Though it’s not really energy and nuclear energy related.) It was this post on uranium chemistry that I found the blog via, and which I found really […]
[…] chemistry for profit and fun. This is quite a cool, impressive example of demonstrating some chemistry in your garage. Informative stuff. I’m impressed that he’s done all that inorganically, with no solvent […]
Useful Information
what is the best method for high carbonate ore , carbonate and bicarbonate with CO2 and o2 is too slow and low recovery , do have type of catalyst can be used to make it faster .
Hi Carl, thanks for an excellent post. How would one get UF3 from UF4, to get a UF4-UF3 salt molten?
http://www.energyfromthorium.com/forum/viewtopic.php?f=6&t=1505
Wow! very interesting and useful to industry. You make it sound simple even though I could never attempt it.
But I wondered if this is a hobby or a business for you. Can you sell this to reactors? The cost of a hobby like this would be huge. Maybe you use a university laboratory. It beats me, anyway
Hi Carl ,
I used to live in Los Angeles,CA but now in Sydney,Australia.What safety clothing,masks etc do you use for this ?
Also,I don’t see ammonium uranyl chloride ? Do you make t and what is it’s modern formula?
Thanks ,
Peter
Wikipedia says that Uranyl peroxide is soluble.
I guess they must be wrong?
Everything is soluble to some degree (equilibrium is described by the solubility constant, Ksp). Uranyl peroxide is practically insoluble in water at neutral pH, or acid hydrogen peroxide solutions down to about pH = 2 in my experience. It will dissolve in carbonate solutions, in very acid solutions, and presumably in other solvents as well.
-Carl
Really interesting stuff, but dare I ask why one needs uranium yellow cake to “serve personal needs.”
The DEA is totally hilarious, The chemicals you could use to make drugs are easily exchangeable, but sometimes legitimate use of those chemicals is necessarily for chemistry. I always made my Iodine by adding KI to HCl making HI. Then I oxidized it with H2O2, and filtered to get semipure elemental I, after that I heated it in a flask connected to a cold finger. After that you get very pure Iodine, a little bit of water might be left on the crystals so a desiccant can be added.
It’s hard to justify manufacturing iodine in light of the fact that it is always available on eBay in reasonable quantities < 250g (per eBay policy). For most legitimate hobby uses 250g is plenty of iodine. In fact, it's the bulk residential manufacture of iodine and other drug precursors that is explicitly not exempt from DEA oversight and can draw interest. My advice is to buy iodine in small quantities on eBay and avoid the various disadvantages–legal, chemical, economic–of cooking it yourself. Whatever you're doing, be careful…
I had an idea, and, being relatively new to chemistry, this may be completely stupid. But it seems to me that an alkali metal could be used in place of calcium. This would result in soluble fluoride salts, making extraction of the pure metal much easier. But I’m probably wrong. It seems that Carl would have thought of this.
Hi First off nice website!!! I picked up a whole bunch of orange thick clumpy powdery rocks thinking its uranium ore, and threw them in a bucket of muriatic acid and after a few days they turned a bright yellow color, is this uranium ore? or am i going in the right direction? or am I lost?thanks awaiting your reply.
Hi Steve, your observations aren’t inconsistent with uranium, but an orange rock forming a yellow solution in HCl is broadly consistent with many other possibilities (e.g. iron). Are there any other reasons to think what you have contains uranium? Tests which would strongly indicate uranium as a major constituent of this rock include radioactivity and UV fluorescence of the HCl solution.
An easy test for uraniferous rock is to wet the surface with dilute nitric or acetic acid, then let it dry. This will convert some of the uranium into uranyl nitrate or uranyl acetate. If, upon irradiation of the treated surface with UV light a green fluorescence is noted, uranium is present. It’s an old miner’s trick that I use to distinguish thorium from uranium in the field.
do you have type of catalyst can be used to make a faster recovery? since carbonate and bicarbonate with CO2 and o2 is too slow and low recovery.
IS THERE ANY WAY TO EXTRACT PEROXIDE FROM URANYL PEROXIDE SURELY A RADICAL METHYL,PHENYL
OR A METAL SODIUM,POTASSIUM OR A QUINONE PERHAPS
P.S SOMETHING TO EXPERIMENT WITH SINCE YOU HAVE THE ABILITY
Do you mean reduce uranyl peroxide to uranium metal? What is the end product you’re after?
The uranyl peroxide in my process is a hydrate of somewhat variable composition, which would likely complicate any attempts to reduce it directly.
Hi Carl, do you remember that have talk about thorium sulfate nonahydrate grown? have send picture via your hotmail, but never have new, so have you received this picture? It was do last mounth. Let me know
Jeff
Do you know if uranium tetrafluoride fluoresces under 365nm UV light.
Thanks
Rotor
I agree with admin, carbonate and bicarbonate with CO2 and o2 is too slow and low recovery. Is there another catalyst you can use?
I don’t know what either of you guys are trying to do with “CO2 and O2.” My procedure never mentions catalysts either. If I had to guess, you are referring to direct oxidation of uranium in your ore into soluble carbonate complexes. This is sometimes done in industry, depending on ore chemistry, and has a reputation for being slow and difficult. But my procedure is an acid leaching procedure…
A question from Brian has still been unanswered and I think it’s an interesting question.
“Really interesting stuff, but dare I ask why one needs uranium yellow cake to ‘serve personal needs.'”
Well, personally, I mostly use refined uranium to do little experiments with fission and fission products. That is to say, I am into it for its nuclear properties. But that’s just me. Other niche uses I’m aware of include DIY glazes for fine art pottery, and staining for amateur electron microscopy. Small-timers have to refine and stockpile their own U because commercial sources tend to have their panties in a bunch and won’t ship to individuals.
Hi Carl,
If I use H2O2 as an oxidizer instead of chlorine bleach will that avoid the production of nasty chlorine gas?
Thanks.
H2O2 doesn’t make as much chlorine as my recommended hypochlorite bleach method of oxidizing the leach, but it still causes a great deal of foaming (oxygen mostly), generates some chlorine gas (i.e. this won’t allow you to bring it indoors), and it’s vastly more expensive. This is why I don’t favor that approach, although it has seen use in industry and I can confirm that it is effective.
hi
very interesting reading good jobb
i live in Norway where we have ca15-25% of the known thorium reserves in a plase caled fenfeltet is just 2h drive from my house it has consentrations of betwen 1-3% Th and 0.7-1%U i m wondering if this prosidure is propriat for Th recovery? or is ther a modification to some part of the prosidure as it is U spesific
brg Thor the viking
This procedure relies on a carbonate-peroxide cycle that is highly selective for uranium. A different chemistry would be required for thorium processing. Thorium is much less amenable to household processing than uranium. I think on the small scale the best approach is probably via fluoride digestion of the ore, but I have not tried any ideas yet. I do have a supply of thorium monazite in preparation for testing some of these ideas.
if you are interrested in trying Th extraction i can send you a sampel as soon as the snow melts. the minneral the Th is in is a charbonatit-hemmatit vulcano chimny caled “rødberg” 1-3%Th
I am interested in going through this process myself. Most of what you have said is detailed well, but I want to know more about safety procedures. I understand that the radiation given off by “yellowcake” can be very dangerous, especially when inhaled. Could you walk us through your process outlining the steps you take for safety at each section? Like: is the ore safe in large quantities? How should you store the ore or the products? If these products are placed too close to each other, could they be dangerous? I would really appreciate your help. I am a young scientist and I am excited to enter the fascinating field of nuclear chemistry, but I want to keep my health! Thanks again!
Ore presents an internal exposure problem due to radon gas. I store it and process it outdoors for this reason.
The liquid and solid wastes from ore processing are highly radioactive and full of toxic metal salts. I dry these wastes in pails and the resulting solid cake is returned to the mine sites and dropped down the shaft. I think this is by far the most responsible way to handle it.
Yellowcake is not particularly hazardous (toxic, radioactive) relative to the ore and the residues. But the hydrogen peroxide used to make it in this process is a strong oxidizer that causes painful burns on contact.
Hope this helps.
[…] irradiated 25 grams of natural uranyl peroxide, freshly prepared from Utah pitchblende ore, overnight with a ~5E+06 n/s PuBe neutron source. This source intensity […]
This is a fine presentation congratulations! But i ADVISE NEVER DO IT if not trained in advanced chemistry and using safety equipmente, because mutch of this is EXTREMLY AZARDOUSE and potential dead poisoning chemicals.
I have some radioactive Fiestaware, so if I could scrape off the protective layer, could I put it into a solvent to avoid scraping all of it off? (to avoid hazardous dust.) Thank you.
Years ago, when chemistry students thought it was funny to blow the hinges of lab doors, it was possible to obtain highly concentrated ammonia (40%?) through local blueprint shops. Iodine crystals were available in 4 OZ bottles through local pharmacist’s.
Reblogged this on Tomás Miguel Roda de Jesus Martins.
thanks for the presentations actually i researching more about U02 Uranium dioxide and i have some glimpse of how it is made but has for me am looking for some who can buy Uranium dioxide cause my brother who is working in uranium mines in DRC cong but he was trained in Israel to work in uranium mine sent me 3kgs of purified Uranium dioxide to sell so am looking for some one who can buy it thanks
Where are you located and what are the specifications of the UO2 your brother sent you?
The DRC’s most famous uranium mine is called Shinkolobwe. It is not too far from Lubumbashi, in Katanga Province. But Shinkolobwe has officially been closed for many years.
-Carl
i really want to know the use of uranium dioxide uo2
Uranium dioxide is the compound used to make most nuclear reactor fuel pellets. It is a semiconductor with some very limited electronics applications.
A most interesting article. I prospected for uranium in Cornwall UK some years ago and collected some 15 lb of Autunite. Having precipitated the sodium duranite from the carbonate I then added to the residue barium nitrate and boiled with nitric acid. To the filtrate I added sulphuric acid to precipitate the sulphates. Saturated sodium hydroxide solution preferentially dissolves out the lead compounds giving a good Pb 212 beta source. This operation was performed in a disposable gloved bag and interestingly every thing with in the bag became radio-active for a few hours. The remaining precipitate was useful as a Radium source.
Martn Page
4-4-2013
hello my name is william I would like to learn if you are a chemistsys also I would like for to re conversate with your self abought chemistry and how to order rock ore by you
Thanks for this. I don’t think I’ll be attempting it for all sorts of reasons but it’s a brilliant read on a subject I think is likely purposely rarely reported/ discussed.
Ok Dumb question and obviously this might not be a great idea… but couldnt you just do a thermite reaction from uo3 + aluminum to get your metal… I understand the reaction is much more violent but I have been pouring iron that way for a bit of time now (for a bit of fun more than anything)
I am on the electronics/programming side of things chemistry is more in the hobby arena for me so I honestly wouldnt mind a bit of feedback here
Carl, I am in the hazardous waste disposal business and I have a question for you, seeing that you are quite knowledgeable in uranium chemistry. A customer of mine has some 100 g of uranyl nitrate. because of the oxidizing capacity it is considered a mixed waste. if I can reduce the nitrate to an oxide, carbonate or other compound, i can save my client thousands of dollars.
Any suggestions?? Thank you,
Hi Ron, uranium can be precipitated from solutions of the nitrate by any alkaline metal hydroxide or by ammonia, forming diuranates generally. These are non-oxidizing yellow to orange-colored solids that can sometimes be difficult to filter from the aqueous waste (containing nitrate anions in this case). As an alternative, you could just send your client’s uranyl nitrate to me. As my blog makes abundantly evident, I collect the hot stuff. I hope this is helpful in some way. Best regards, Carl
Thanks Carl for the very quick response. I appreciate the simple answer. I would be solidifying the resulting waste for landfill in the end.
As far as sending it to you, I would have to check with the folks that have it (and they just happen to be in NM, where I lived for 15 years before coming to SC.) You know how people are scared to death of liability these days.
In the event that they will take you up on the offer, here is my email address. send me a note with your contact info and I will have it on hand. Thanks again.
r o n (at)echelonenvironmental.net
Reblogged this on Chirpy's Blog.
Hi Carl,
Nice to see this detailed explanation on U extraction publish it online. From where I come from it is considered illegal to have a Geiger counter without a permit and here you reduce uranium.
Lucky you!
Can you tell me witch contry forbids geiger counters?
Hi Thor
Romania.
thanks for the cake NOT!.
love you
Does one need a licence from the NRC in order to collect the uranium ore? To synthesize uranium compounds? To purify and producing uranium metal?
According to 10 CFR Chapter 1 Part 40 Section 13, I am exempt from the regulations in that part if I just want to collect unrefined, unprocessed ore. However I do not clearly understand who can receive a general license for small quantities of source materials. Paragraph “a” of 10 CFR Part 40 Section 22 states that the general license is given to firms, institutions, agencies. Does that mean an individual needs to be part of a firm, institution, or agency or does that mean that an individual cannot receive a general license for small quantities of source material? If I understand this correctly than ANY firm, institution, or government agency can say they have a general license? (so Burger King or McDonalds can legally obtain 1.5 kg of uranium if they wanted?)
Very interesting article! Could you please advise on the preparation of uranyl nitrate from uranium metal? I’ve tried dissolving U metal in concentrated nitric acid, and I’m getting a yellow-tinted solution. I’m not sure that it’s uranyl nitrate though, especially because I couldnt get it to recrystallize in the freezer or precipitate it out using ammonium hydroxide? Is there any other specific process?
Uranium dissolved in nitric acid will produce uranyl nitrate, and GENTLE evaporation (preferably under vacuum) will yield crystals of uranyl nitrate hexahydrate. Overheating decomposes uranyl nitrate, so you must be careful to keep the solution cool as it evaporates. Ammonium hydroxide should precipitate even fairly dilute uranyl ions as ADU at a pH of 8 or higher. However, be careful to exclude carbonate or sources of CO2 since they will complex uranium back into solution. It may be hard to tell you have a precipitate at low concentrations because the precipitate tends to be very fine and doesn’t settle readily. Good luck!
Thanks! I’ll try that out!
Wow, this is really in depth. Got a question though. If one obtained several liters (10-15) of D2O and Scientific grade graphite (as a reflector/control rods) could you dissolve Uranium nitrate in the D2O and use it to build a small aqueous style reactor like the ones oak ridge bulit? Say the size of a five gallon bucket? (NOT IN ONE)
If so how small could you build one (amount of nitrate needed in grams) and what would you need to build the physical container from in order to ward off the corrosive nature of the aqueous fuel?
Would building a fusor based neutron source to “cold start” it reduce the fuel I would need?
The general equation for the radius of a homogeneous reactor R = pi (M)^1/2/(k -1)^1/2 where M = L^2 + Ls^2. L is the diffusion length = Lt/3Za. Lt is the transport length for D it is about 2.52 cm and Za the abortion cross-section, principally due to D and about 0.002 barns. For natural uranium N238 to N235 of 140 : 1 Kinf. is about 1.15 using D2O. Putting these together gives a radius of about 2 meters and a suitable reflector would reduce this a bit. If you could get a few handfuls of U235 to sprinkle in it, then it might be reduced to a few gallons in size.
I don’t believe it is possible to make an aqueous homogeneous critical assembly from nat-U uranyl nitrate and heavy water, in any concentration or size. A quick MCNP perturbation study shows you can come pretty close (k_inf ~ 0.85), but there is too much non-fission capture on U-238 unless you consider inhomogeneous uranium distributions akin to what the Germans were doing in WW2. The other problem here is that uranyl nitrate is always a hydrate (hexahydrate, specifically) in its stable, manufactured form. To use natural-isotopic uranyl nitrate successfully in a critical assembly, light hydrogen would have to be removed from this material either by desiccation (very dangerous in quantity, this is an explosive salt) or by preparation from D2O chemistry. Finally, the presence of a neutron source like a fusor (or any other source) would not change the criticality condition, which is solely a function of materials and geometry. However, you could use such a source to increase neutron population and generated power via subcritical multiplication.
For natural uranium UO2-H2O homogenous system the maximum Kinfn is 0.8 at a ratio of H-U of between 5 to10. This factor can only be raised by enriching to 3% U235 as occurred some 2000 million years ago ( Oklo) and predicted by Kazuo Kuroda. For a D2O- UO2 system the maximum Kinfin is 1.17 for a D-U of between 300 to 400. The abortion cross section for D is 0.00046 b and that of H 0.332 b, heavy water is usually 99.75% D2O and 0.25 H2O so the effective value is 0.00259 b.
The only time that I am aware of natural uranium dissolved in D2O was tried was in 1940 when 112 litres of heavy water, ordered by France, was smuggled across the north sea by fishing boat from Norway. At Cambridge laboratory a slurry of U3O8 was created inside a 60 cm aluminium sphere and immersed in mineral oil. There was not enough material to produce a sustained chain reaction but the neutron decay of injected neutron yielded an experimental K factor. At the time Britain stood alone in the world against Germany and for security reasons the 112 litres of heavy water was shipped to Canada which led eventually to the development of the CANDU reactors.
For a uranium-graphite system the max. K is 0.81 at a C-U ratio of about 400and this can be improved by adopting a heterogeneous arrangement. This is achieved because the MFP for fast neutrons in natural uranium is about 3 cm but that of thermal about 1.3 cm. By making the uranium slug about 2.5 cm in dia. and spacing them at one slowing down distance about 19 cm the factor K can be raised above 1. Such a reactor is about 10 or more metres in dia.
Could you direct me to where I can get a copy of the MCNP software and a tutorial on how to use it?
Hi David, you must obtain MCNP from RSICC: https://rsicc.ornl.gov/codes/bcc/bcc0/bcc-009.html The code’s price and availability depend on review of your request for export control and nonproliferation compliance. I think you can get the executable-only package for $150 if a US citizen. If you want the full source code, you may have to wait a long time while your information is screened by the USDOE and they may charge you more. I am happy to share the input “deck” for the criticality problem referenced earlier if you get the code and want to play around with it.
Homogeneous reactor.
For a homogeneous configuration the fast fission factor ‘e’ is close to unity but by lumping the fuel this can be raised to 1.1 or so .The number of fast fission neutrons produced per thermal neutron absorbed in uranium ‘z’ is for a U28 / U25 of 137.8 is 1.335.
Using the four factor equation for Kinf, the condition that a chain reaction be possible in a homogeneous system of finite size is such that Kfin >1, or zfp >1, that is pf >1/z = 0.75, where ‘f’ = thermal utilization and ‘p’ = resonance escape probability.
For this the resonance escape probability ‘p’ and the thermal utilization ‘f’ must both be in the region of 0.85. Setting the moderator to fuel as N1/No, an analysis gives:-
For U-Graphite pf = 0.604 and Kinf 0.81 at N1/No = 400.
UO2-H2O__ pf = 0.595__Kinf 0.8___5.
UO2-D2O___ pf = 0.874__Kinf 1.17__300-400.
Hence a chain reaction is a homogeneous system is only possible for the D2O-UO2 arrangement.
As far as I am aware nobody has ever built such an arrangement. The big problem would be the radiological-decomposition of the moderator which for enriched systems was overcome by using a catalytic converter.
I would be interested in the results of your computer programme.
Hi Martin,
I’d be happy to share outputs from MCNP with you, but have no way of getting in touch. Shoot me an email and I will reply. The real problem with uranyl nitrate, as I mentioned, is that it is always crystallized as a hexahydrate commercially, and even this little bit of light hydrogen is a huge reactivity penalty in nat-U/D2O homogenous reactors. That alone is a good reason to choose something else (like the oxide, as you mentioned).
-Carl
What is needed is a compound of uranium that has a low neutron absorption and is soluble in water. Halban and Kowarski used a slurry of uranium oxide which I assume needed constant agitation, probably had a dispensable first year undergraduate to stir it!
Contact tangazazen@aol.com
Foot note: Uranium nitrate might not be the best solution, nitrogen absorption cross section a little too large at 1.9 barns and uranium sulphate only a little better (Sulphur 0.5 b). Best would be uranium oxide (Oxygen 0.00027 b).
Foot note: Halban and Kowarski at the Cavendish Laboratory in England in December 1940, from experiments with 112 litres of heavy water and a slurry of U3O8 determined the multiplication factor Kinf of 1.18+-0.07 for a ratio of deuterium to uranium of 380 to 1, and 1.09+-0.03 for D/U of 160 to 1.
Just a foot note.
The effect of enrichment can be judged by the Tokaimura accident. The uranium nitrate solution was 40 gm/100cc enriched to 18.8 % U235 (total 16 Kg U). The container which was externally water cooled (neutron reflector) was 45 cm dia. The solution was poured in until the depth was 25 cm (40 litres) which at this point it went critical. For a depth equal to the diameter, criticality would occur with a smaller amount of the solution (optimum geometry). This accident resulted in the death of the two operator.
The separation of Lead 210.
The starting material (about 14 lb) was a secondary mineral Autunite, a calcium uranyl phosphate from near Ponsaooth, a small village in Cornwall. The ground up material was roasted and boiled with sodium carbonate. After filtering the ph. was raised with sodium hydroxide to precipitate the sodium duranate.
To the mineral residue was added a few grams of barium nitrate and boiled with dilute nitric acid. This was filtered a couple of times to remove the Iron hydroxide. The addition of sulphuric acid precipitated the sulphates, principally those of barium.
A saturated solution of Sodium Hydroxide has the property of preferentially dissolving the Lead sulphate and which was then separated by filtration. The addition of sulphuric acid to the sodium/lead sulphate precipitated the PbSO4. The mineral came from an area that was long ago well known for lead mining and so contained a considerable amount of inert lead.
With an end window Geiger tube and aluminium filters, the radio-activity was determine to be beta. The count rate versus mg/cm2 was plotted using the K. Otazai and S. Hyashi beta plot and gave an energy of 1.15 Mev close to the published figure on 1.161 Mev.
Wow, thanks.
Neutrons ?
As a foot note; the residual barium sulphate of course also contain a ‘speck’ of radium sulphate, enough to over excite the end window Geiger counter. This was mixed with beryllium hydroxide derived from some old NaI x-ray scintillators windows.
A boron10 lined neutron detector was mounted axially in a wax filled one gallon paint can and a back ground count of 90 per 30 minutes was measured. The radium-beryllium mixture was covered with 2mm Pb as the neutron counter had a small response to gamma rays. When the Ra-Be is laid directly against the neutron detector without the wax container, there was no change in the background count. But with the detector in the wax container and the Ra-Be place radially in the wax, the count rose to 150 per 30 minutes.
Very interesting!
Few month ago, i read an old document from 1961
it said Uranium metal can be produced by reacting uranium hexafluoride and sodium metal.
Which produce uranium metal and sodium fluoride.
After the reaction, sodium fluoride can be removed by dissolving reaction product in water.
I wonder if it is possible with uranium tetrafluoride.
This is brilliant. I have just produced uranium peroxide and sodium diuranate from this. They will make handsome additions to my radioactive collection. Thank you for publishing this.
With reference to the discovery and isolation of potassium from potash,1800’s elemental chemistry. I did not watch the video but would keenly zap anything, i just had one look at the final product and knew it could be retrieved through electrolysis.
I have noted how clinical the commentary is but i have to further that 10 Amps is enough to light up old sparky.
Hello Carl,
In Europe, Hydrogen peroxide is not available in 27%, but only in 12%. Would that 12% H2O2 work in Part IV of your process?
Thanks,
Chriss
Indonesian uranium:
– Liquid 10.5kg more + 2kg paste (53.46%) Liquid? Jelly?
– Metal plate 1.552gr (88.34%) Metal plate?
– Pecok metal 2.256gr (82.06%) Metal Pecok?
– Metal plate 2,900gr (not yet in the lab) is estimated at 88% and above
TOTAL 19.200kg
[…] Uranium Chemistry (2008) 12 by ohaikbai | 1 comments on Hacker News. […]
[…] Uranium Chemistry (2008) 12 by ohaikbai | 1 comments on Hacker News. […]
[…] Article URL: https://carlwillis.wordpress.com/2008/02/20/uranium-chemistry/ […]
[…] https://carlwillis.wordpress.com/2008/02/20/uranium-chemistry/ […]